How to represent a protein sequence
September 29, 2023
In the last decade, innovations in DNA sequencing propelled biology into a new information age. This came with a happy conundrum: we now have many orders of magnitude more protein sequences than structural or functional data. We uncovered massive tomes written in nature's language – the blueprint of our wondrous biological tapestry – but lack the ability to understand them.
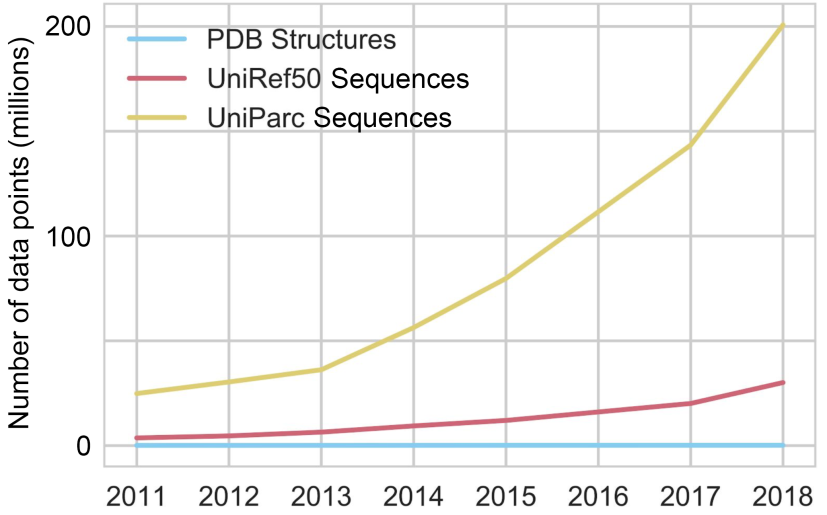
The red and yellow lines represent the number of available sequences in public online databases; the blue line represents the number of available structures, whose increase is unnoticeable in comparison. Figure from 1.
An important piece of the puzzle is the ability to predict the structure and function of a protein from its sequence.
In this case, structural or functional data are labels. In supervised learning, we would show our model many sequences and iteratively correct its predictions based on how closely they match the corresponding, expected labels.
When labels are rare, as in our case with proteins, we need to rely on more unsupervised approaches like this:
-
Come up with a vector representation of the protein sequence that captures its important features. The vectors are called contextualized embeddings. This is no easy task: it's where the heavy lifting happens and will be the subject of this post.
Representation vectors are created from the amino acid sequence. Each vector corresponds to an amino acid (hover to view). The values in the vectors are made up. The length of each vector is typically between several hundred to a few thousand.
-
Use the representation vectors as input to some supervised learning model. The information-rich representation has hopefully made this easier that 1) we don't need as much labeled data and 2) the model we use can be simpler, such as linear or logistic regression.
This is referred to as transfer learning: the knowledge learned by the representation (1.) is later transferred to a supervised task (2.).
What about MSAs?
We talked in a previous post about ways to leverage the information hidden in Multiple Sequence Alignments (MSAs): the co-evolutionary data of proteins.
MSA
An MSA contains different variants of a sequence. The structure sketches how the amino acid chain might fold in space (try dragging the nodes). Hover over each row in the MSA to see the corresponding amino acid in the folded structure. Hover over the blue link to highlight the contacting positions.
We talked about robust statistical models that accomplish:
However, those techniques don't work well on proteins that are rare in nature or designed de novo, where we don't have enough co-evolutionary data to construct a good MSA. In those cases, can we still make reasonable predictions based on a single amino acid sequence?
One way to look at the models in this post is that they are answers to that question, picking up where MSAs fail. Moreover, models that don't rely on MSAs aren't limited to a single protein family: they understand some fundamental properties of all proteins. Beyond utility, they offer a window into how proteins work on an abstraction level higher than physics – on the level of manipulatable parts and interactions – akin to linguistics.
Representation learning
The general problem of converting some data into a vector representation is called representation learning, an important technique in natural language processing (NLP). In the context of proteins, we're looking for a function, an encoder, that takes an amino acid sequence and outputs a bunch of representation vectors.
An encoder converts a sequence into representation vectors (hover to view). The length of each vector is typically between several hundred to a few thousand.
Tokens
In NLP lingo, each amino acid is a token. An English sentence can be represented in the same way, using characters as tokens.
Hover to view the representation vector of each character token.
As an aside, words are also a reasonable choice for tokens in natural language.
Hover to view the representation vector of each word token.
Current state-of-the-art language models use something in-between the two: sub-word tokens. tiktoken is the tokenizer used by OpenAI to break sentences down into lists of sub-word tokens.
Context matters
If you are familiar with NLP embedding models like word2vec, the word embedding might be a bit confusing. Vanilla embeddings – like the simplest one-hot encodings or vectors created by word2vec – map each token to a unique vector. They are easy to create and often serve as input to neural networks, which only understand numbers, not text.
In contrast, our contextualized embedding vectors for each token, as the name suggests, incorporates context from its surrounding tokens. Therefore, two identical tokens don't necessarily have the same contextualized embedding vector. These vectors are the output of our neural networks. (For this reason, I'll refer to these contextualized embedding vectors as representation vectors – or simply representations.)
As a result of the rich contextual information, when we need one vector that describes the entire sequence – instead of a vector for each amino acid – we can simply average the values in each vector.
Now, let's work on creating these representation vectors!
Creating a task
Remember, we are constructing these vectors purely from sequences in an unsupervised setting. Without labels, how do we even know if our representation is any good? It would be nice to have some task: an objective that our model can work towards, along with a scoring function that tells us how it's doing.
Let's come up with a task: given the sequence with some random positions masked away
which amino acids should go in the masked positions?
We know the ground truth label from the original sequence, which we can use to guide the model like we would in supervised learning. Presumably, if our model becomes good at predicting the masked amino acids, it must have learned something meaningful about the intricate dynamics within the protein.
This lets us take advantage of the wealth of known sequences, each of which is now a labeled training example. In NLP, this approach is called masked language modeling (MLM), a form of self-supervised learning.
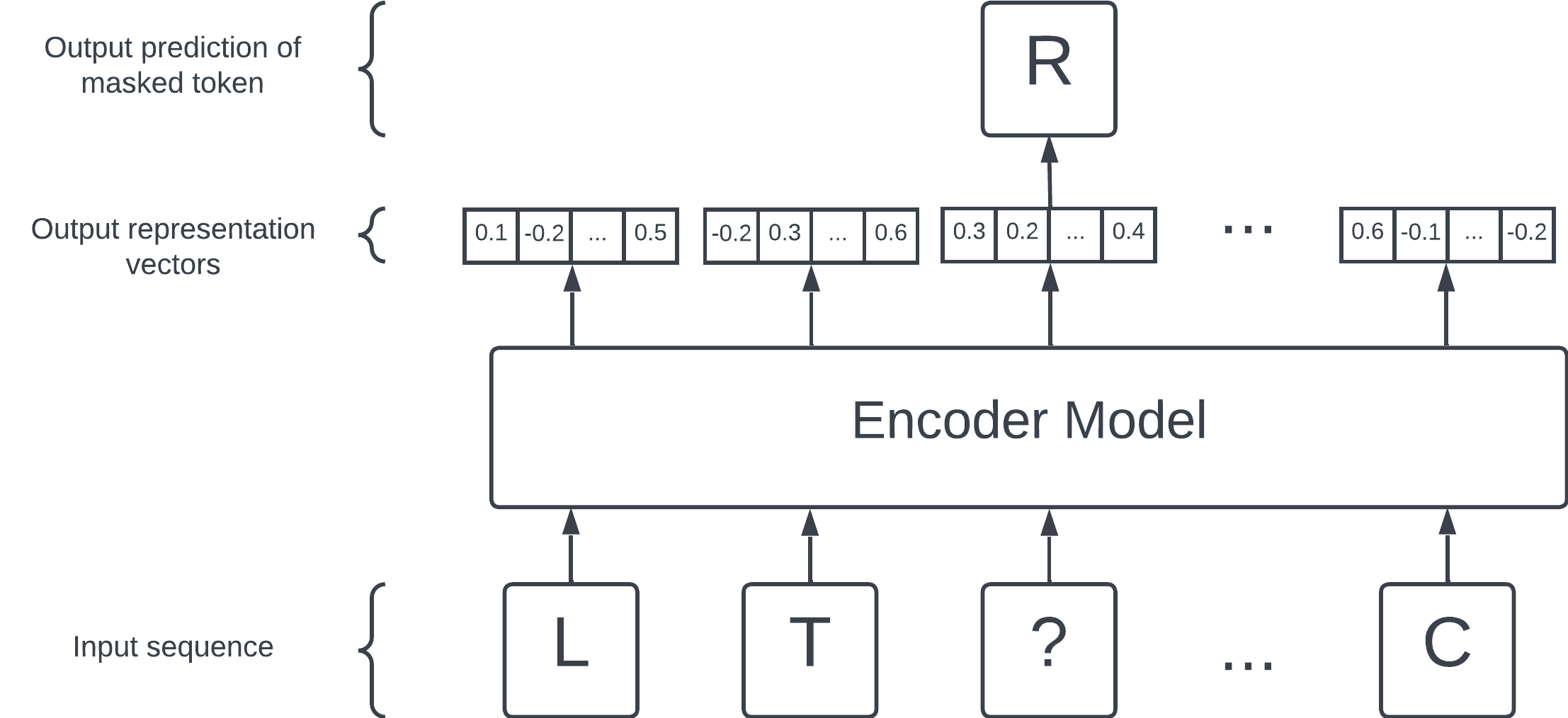
The masked language modelling objective. Hide a token (in this case, R) and ask the encoder model to predict the hidden token. The encoder model is set up so that, while attempting and learning this prediction task, representation vectors are generated as a side effect.
Though we will focus on masked language modeling in this post, another way to construct this self-supervision task is via causal language modeling: given some tokens, ask the model to predict the next one. This is the approach used in OpenAI's GPT.
The model
(This section requires some basic knowledge of deep learning. If you are new to deep learning, I can't recommend enough Andrej Karpathy's YouTube series on NLP, which starts from the foundations of neural networks and builds to cutting-edge language models like GPT.)
The first protein language encoder of this kind is UniRep (universal representation), which used a technique called Long Short Term Memory (LSTM) 1. (It uses the causal instead of masked language modeling objective, predicting amino acids from left to right.)
More recently, Transformer models that rely on a mechanism called self- attention have taken the spotlight 5. BERT stands for Bidirectional Encoder Representations from Transformer and is a state-of-the-art natural language encoder developed at Google 2. We'll focus on a BERT-like encoder model applied to proteins.
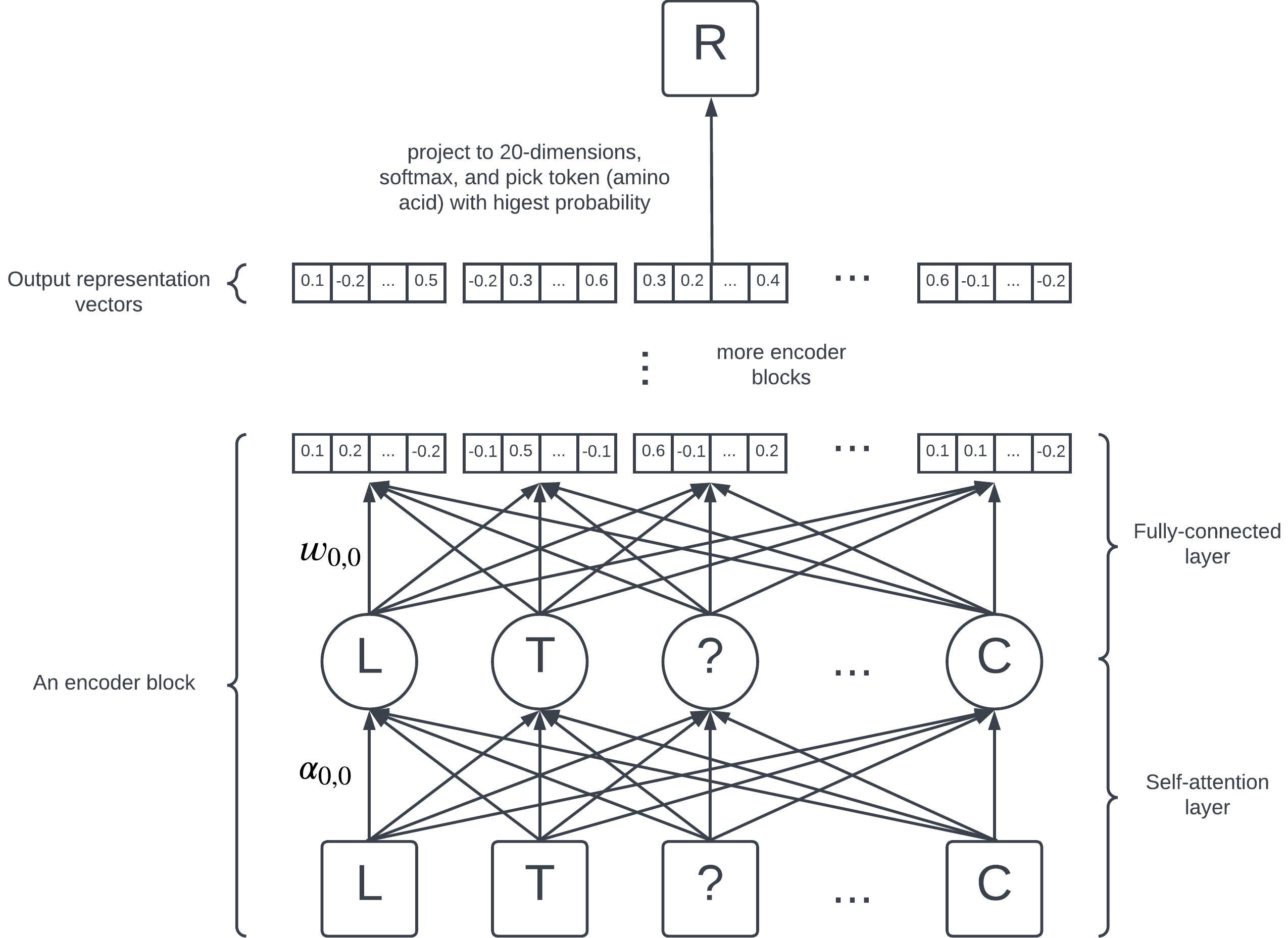
A simplified diagram of BERT's architecture.
BERT consists of 12 encoder blocks, each containing a self-attention layer and a fully-connected layer. On the highest level, they are just a collection of numbers (parameters) learned by the model; each edge in the diagram represents a parameter.
Roughly speaking, the parameters in the self-attention layer (also known as attention scores) capture the alignment, or similarity, between two amino acids. If is large, we say that the token attends to the token. Intuitively, token is "interested" in the information contained in token , presumably because they have some relationship. Exactly what this relationship is might not be known, or even understandable, by us: such is the power – as well as peril – of the attention mechanism. Throughout the self-attention layer, each token can attend to different parts of the sequence, focusing on what's relevant to it and glancing over what's not.
Here's an example of attention scores of a transformer trained on a word-tokenized sentence:
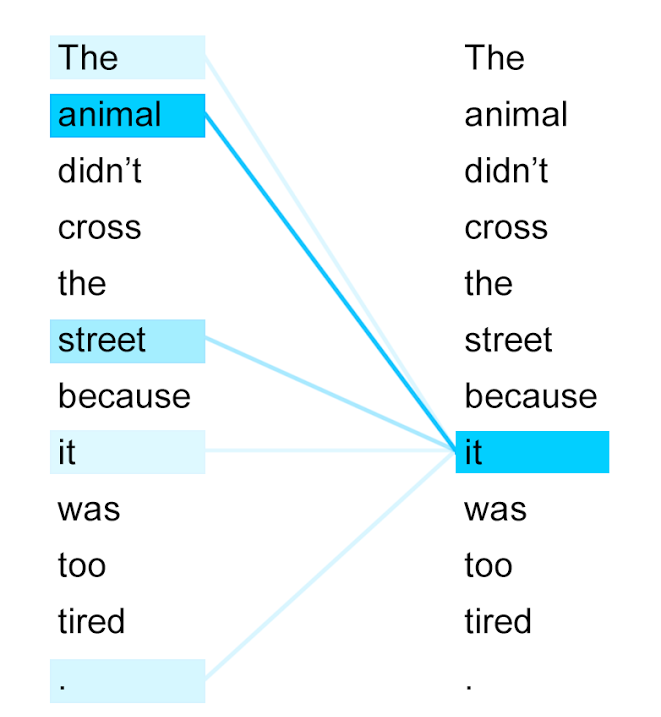
Self-attention visualization of a word-tokenized sentence. Deeper blue indicates higher attention score.
The token "it" attends strongly the token "animal" because of their close relationship – they refer to the same thing – whereas most other tokens are ignored. Our goal is to tease out similar semantic relationships between amino acids.
The details of how these attention scores are calculated are explained and visualized in Jay Alammar's amazing post The Illustrated Transformer. Here's a helpful explanation on how they differ from the weights in the fully-connected layer.
As it turns out, once we train our model on the masked language modeling objective, the output vectors in the final layers become informative encodings of the underlying sequence – exactly the representation we've set out to build.
There are more details
I hoped to convey some basic intuition about self-attention and masked language modeling and have of course left out many details. There's a short list:
-
The attention computations are usually repeated many times independently and in parallel. Each layer in the neural net contains sets of attention scores, i.e. attention heads ( in BERT). The attention scores from the different heads are combined via a learned linear projection 5.
-
The tokens first need to be converted into vectors before they can be processed by the neural net.
- For this we use a vanilla embedding of amino acids – like one-hot encoding – not to be confused with the contextualized embeddings that we output.
- This input embedding contains a few other pieces of information, such as the positions of each amino acid within the sequence.
-
Following the original Transformer, BERT uses layer normalization, a technique that makes training deep neural nets easier.
-
There are 2 fully-connected layers in each encoder block instead of the 1 shown in the diagram above.
Using the representation
Once we have our representation vectors, we can train simple models like logistic regression with our vectors as input. This is the approach used in ESM, achieving state-of-the-art performance on predictions of 3D contacts and mutation effects 3 4. We can think of the logistic regression model as merely teasing out the information already contained in the input representation, an easy task. (We're omitting a lot of details, but if you're interested, please check out those papers!)
We saw in the previous post that with clever samplings approaches like Markov Chain Monte Carlo (MCMC), a good predictive model can be used to generate new sequences. That's exactly the approach taken by researches from the Church lab leveraging UniRep for protein engineering 6:
Start with UniRep, which takes in a protein sequence and outputs a representation vector. UniRep is trained on a large public sequence database called UniRef50.
Fine-tune UniRep by further training it on sequences from the target protein's family, enhancing it by incorporating evolutionary signals usually obtained from MSAs.
Experimentally test a small number of mutants (tens) and fit a linear regression model on top of UniRep's representation to predict performance given a sequence.
Propose various mutants and ask the linear regression model to evaluate them, all in silico. Apply the Metropolis-Hastings acceptance criterion repeatedly to generate a new, optimized sequence. (If this sounds unfamiliar, check out the previous post!)
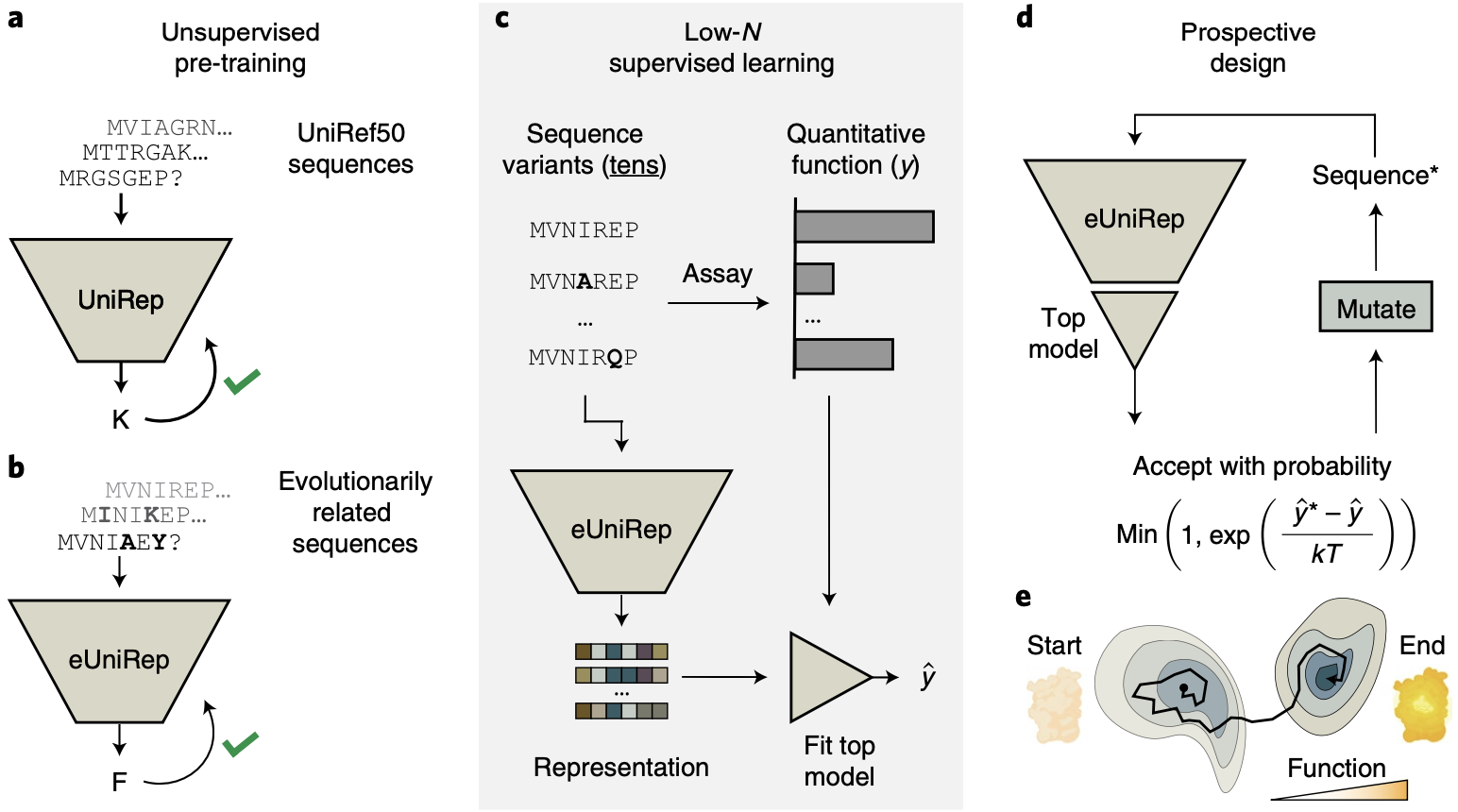
Protein engineering with UniRep. This process is analogous to to meandering the sparsely functional sequence space in a guided way (e). Figure from 6.
A peek into the black box
We've been talking a lot about all this "information" learned by our representations. What exactly does it look like?
UniRep
UniRep vectors capture biochemical properties of amino acids and phylogeny in sequences from different organisms.
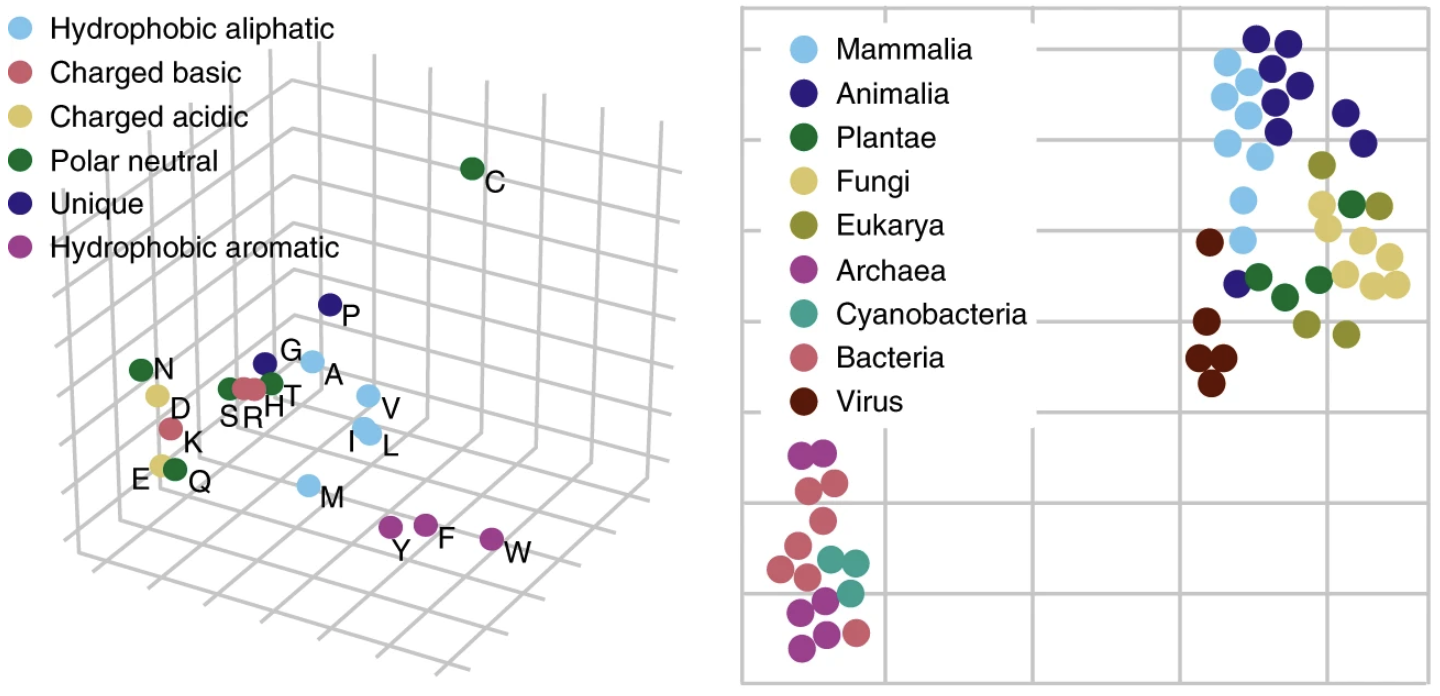
(Left) Feed a single amino acid into UniRep and take the output representation vector. Applying PCA and plotting the representation vector obtained for each amino acid along the top 3 principle components, we see a clustering by biochemical properties. (Right) For an organism, take all of its protein sequences (its proteome), feed each one into UniRep, and average over all of them to obtain a proteome-average representation vector. Applying t-SNE to visualize these vectors in 2-dimensions, we see a clustering by phylogeny. Figure from 1.
More incredibly, one of the neurons in UniRep's LSTM network showed firing patterns highly correlated with the secondary structure of the protein: alpha helices and beta sheets. UniRep has clearly learned meaningful signals about the protein's folded structure.
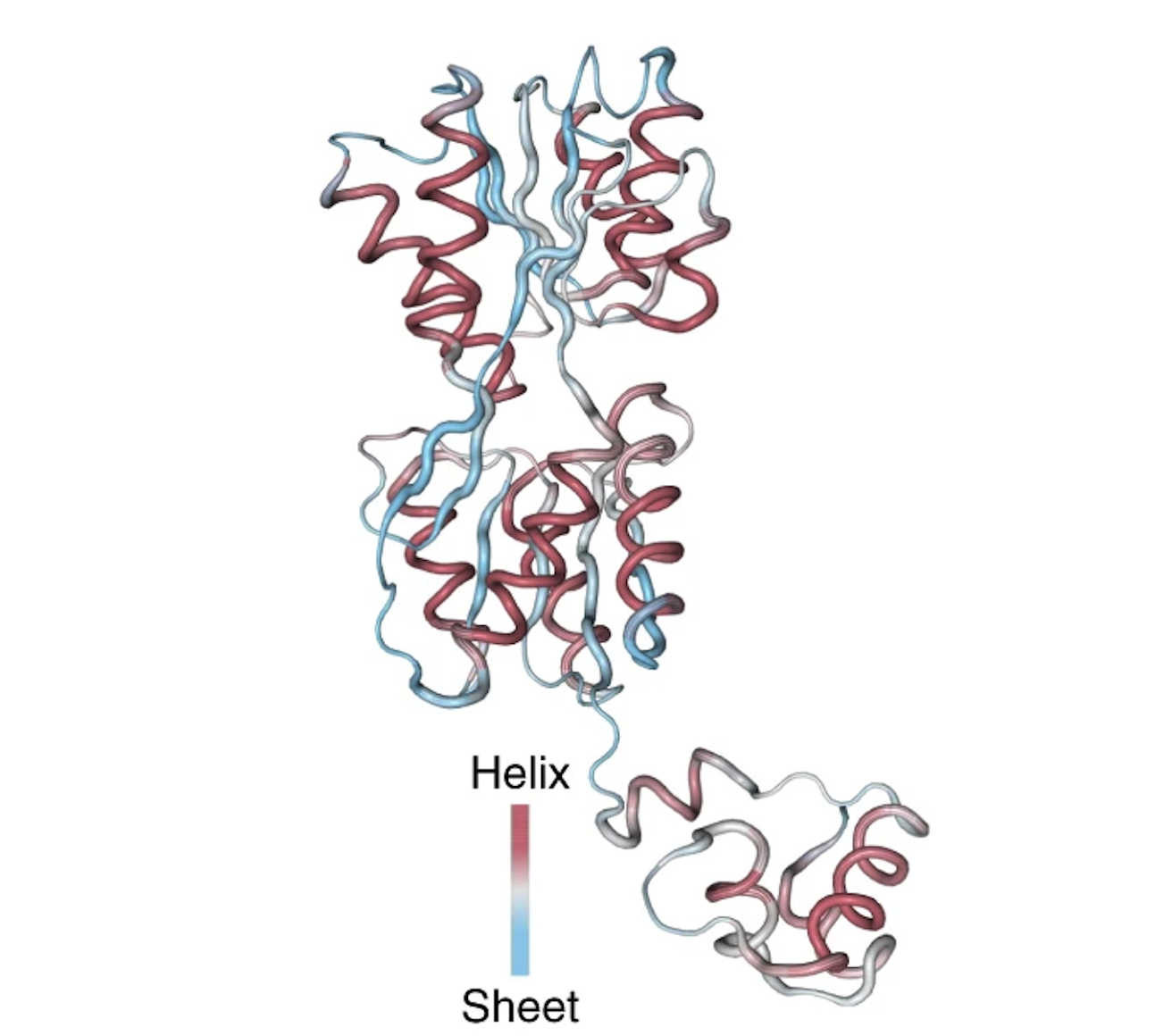
The activations of the neuron are overlaid with the 3D structure of the Lac repressor protein. The neuron has high positive activations at positions that correspond to an alpha helix, and high negative activations at positions that correspond to an beta sheet. Figure from 1.
Transformer models
In NLP, the attention scores in Transformer models tend to relate to the semantic structure of sentences. Does attention in our protein language models also capture something meaningful?
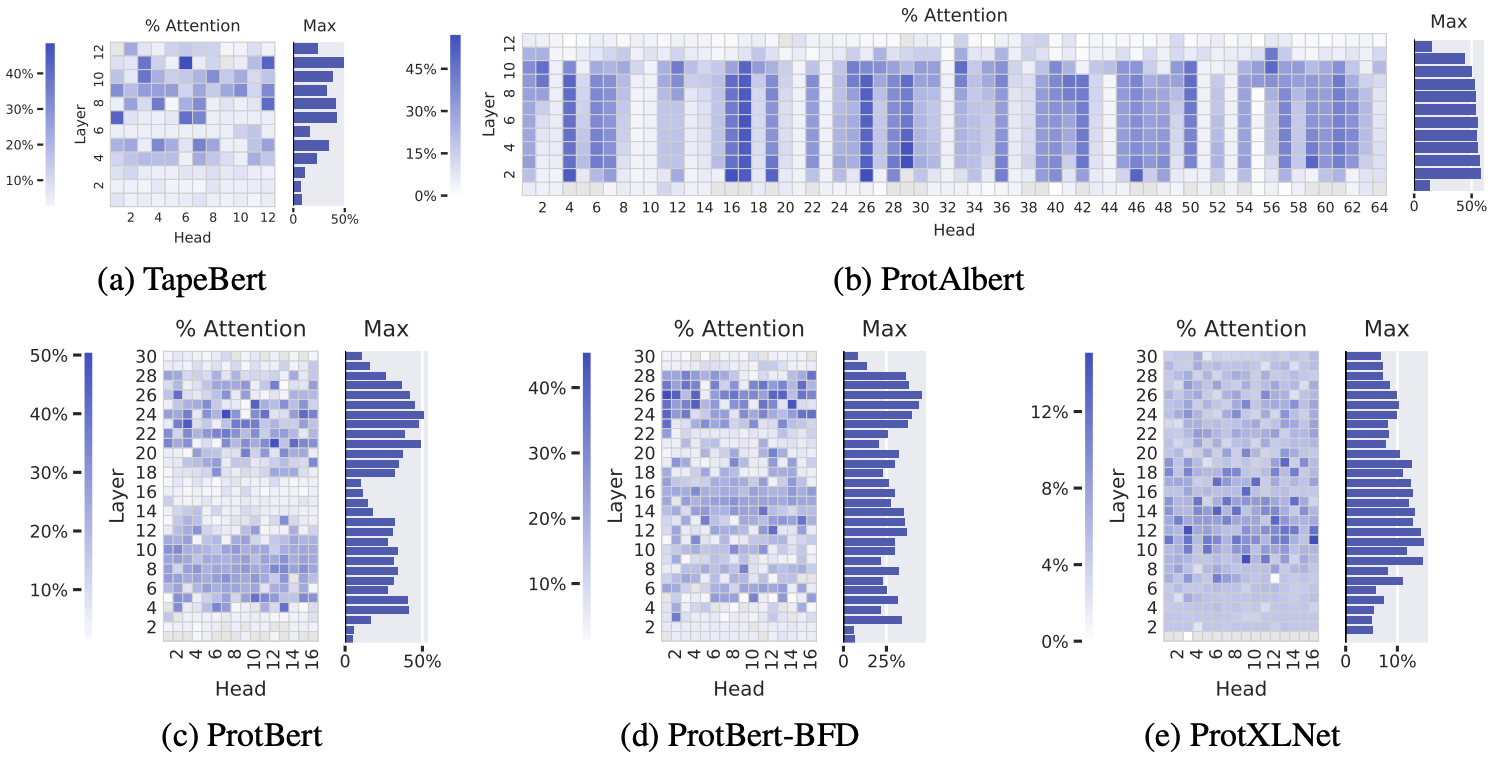
Let's look at 5 unsupervised Transformer models on proteins sequences – all trained in the same BERT-inspired way we described 7. Amino acid pairs that with high attention scores are more often in 3D contact in the folded structure, especially in the deeper layers.
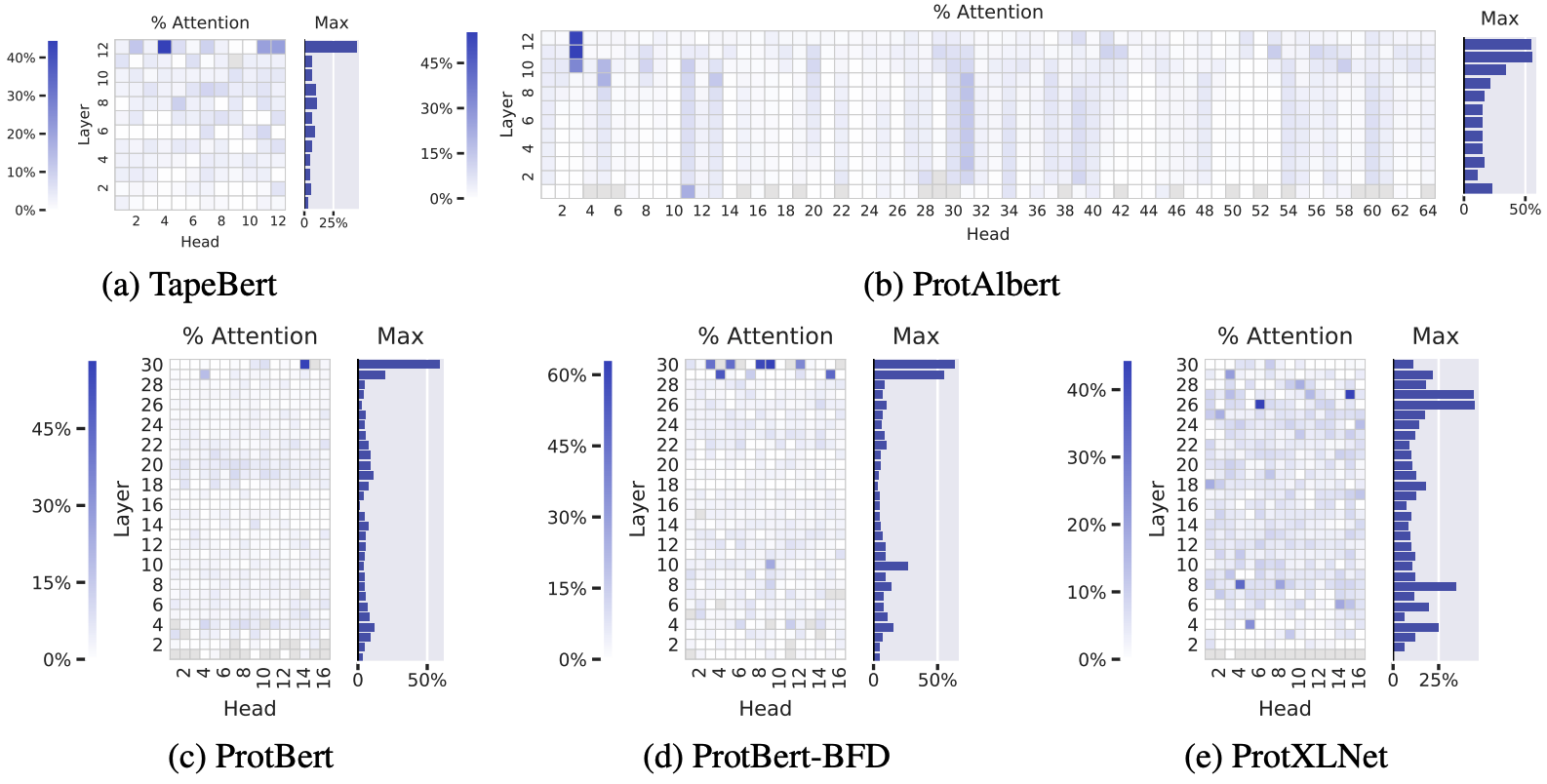
The percentage of high-confidence attention scores that correspond to amino acids positions in 3D contact. Deeper blue reflects higher correlation between attention scores and contacts. Data is shown for each attention head in each layer, across 5 BERT-like protein language models. Figure from 7.
Similarly, a lot of attention is directed to binding sites – the functionally most important regions of a protein – throughout the layers.
The percentage of high-confidence attention scores that correspond to binding sites. These are positions j part of binding sites that have high , i.e. positions that have attention directed to them. Figure from 7.
Applying supervised learning to attention scores – instead of output representations – also achieves astonishing performance in contact prediction. Compared to GREMLIN, an MSA-based method similar to the one we talked about in the previous post, logistic regression trained on ESM's attention scores yielded better performance after only seeing 20 (!) labeled training examples.
Further reading
I recommend Jay Alammar's post on encoder models like BERT and Mohammed AlQuraishi's post on the importance of unsupervised learning in protein science.
References
Alley, E.C. et al. Unified rational protein engineering with sequence-based deep representation learning. Nat Methods 16, 1315–1322 (2019).
Delvin, J. et al. BERT: Pre-training of deep bidirectional transformers for language understanding. arXiv (2018).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. (2021).
Rao, R.M. et al. Transformer protein language models are unsupervised structure learners. Science. 2020;369(6502):440–445 (2020).
Vaswani A. et al. Attention is all you need. NeurIPS (2017).
Biswas, S. et al. Low-N protein engineering with data-efficient deep learning. Nat Methods 18, 389–396 (2021).
Vig. J. et al. BERTology Meets Biology: Interpreting Attention in Protein Language Models. arXiv (2020).
Alley, E.C. et al. Unified rational protein engineering with sequence-based deep representation learning. Nat Methods 16, 1315–1322 (2019).
Delvin, J. et al. BERT: Pre-training of deep bidirectional transformers for language understanding. arXiv (2018).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. (2021).
Rao, R.M. et al. Transformer protein language models are unsupervised structure learners. Science. 2020;369(6502):440–445 (2020).
Vaswani A. et al. Attention is all you need. NeurIPS (2017).
Biswas, S. et al. Low-N protein engineering with data-efficient deep learning. Nat Methods 18, 389–396 (2021).
Vig. J. et al. BERTology Meets Biology: Interpreting Attention in Protein Language Models. arXiv (2020).